


Cefuroxime sodium for injection 750mg
Summary of Product Characteristics Updated 20-Aug-2018 | Flynn Pharma Ltd
- Name of the medicinal product
Cefuroxime sodium for injection 750mg - Qualitative and quantitative composition
Each vial contains, as the active ingredient, cefuroxime sodium for injection equivalent to 750mg of cefuroxime.
Excipients with known effects:
Each vial contains 40.6 mg sodium.
For the full list of excipients, see section 6.1. - Pharmaceutical form
Vials containing an off-white to slightly yellow sterile powder for solution for injection or infusion. - Clinical particulars
4.1 Therapeutic indications
Cefuroxime sodium for injection is indicated for the treatment of infections listed below in adults and children, including neonates (from birth) (see sections 4.4 and 5.1).
• Community acquired pneumonia
• Acute exacerbation of chronic bronchitis
• Complicated urinary tract infections, including pyelonephritis
• Soft-tissue infections: cellulitis, erysipelas and wound infections
• Intra-abdominal infections (see section 4.4)
• Prophylaxis against infection in gastrointestinal (including esophageal), orthopedic, cardiovascular, and gynecological surgery (including cesarean section)
In the treatment and prevention of infections in which it is very likely that anaerobic organisms will be encountered, cefuroxime should be administered with additional appropriate antibacterial agents.
Consideration should be given to official guidance on the appropriate use of antibacterial agents.
4.2 Posology and method of administration
Posology
Table 1. Adults and children ≥ 40 kg
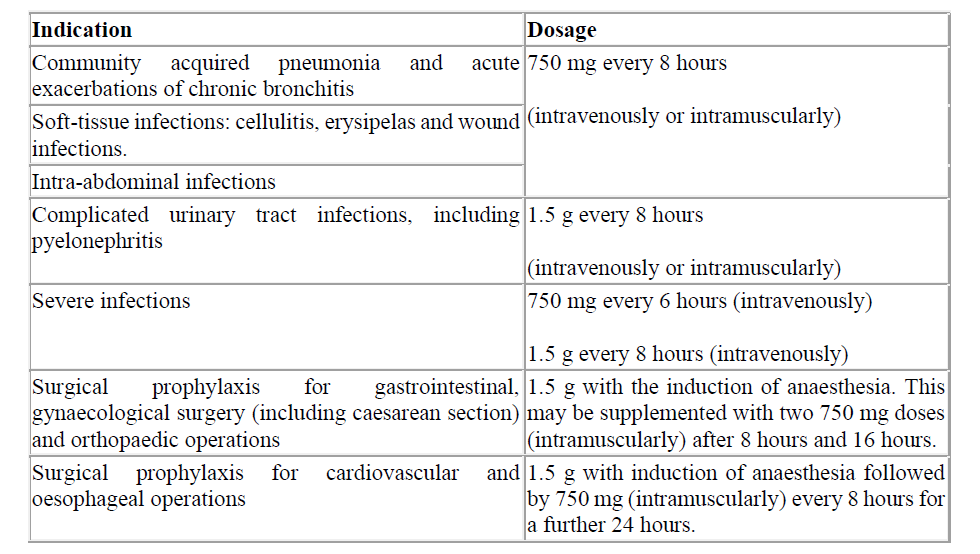
Table 2.Children < 40 kg

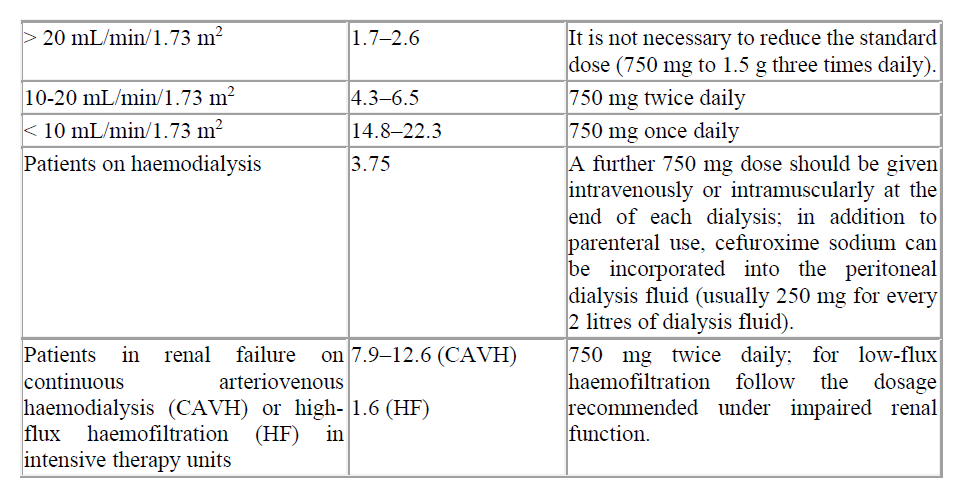
Renal impairment
Cefuroxime is primarily excreted by the kidneys. Therefore, as with all such antibiotics, in patients with markedly impaired renal function it is recommended that the dosage of Cefuroxime should be reduced to compensate for its slower excretion.
Table 3. Recommended doses for Cefuroxime in renal impairment

Hepatic impairment
Cefuroxime is primarily eliminated by the kidney. In patients with hepatic dysfunction this is not expected to effect the pharmacokinetics of cefuroxime.
Method of administration
Cefuroxime should be administered by intravenous injection over a period of 3 to 5 minutes directly into a vein or via a drip tube or infusion over 30 to 60 minutes, or by deep intramuscular injection. Intramuscular injections should be injected well within the bulk of a relatively large muscle and not more than 750 mg should be injected at one site. For doses greater than 1.5 g intravenous administration should be used. For instructions on reconstitution of the medicinal product before administration, see section 6.6.
750 mg powder for solution for infusion.
For instructions on preparation of the medicinal product before administration, see section 6.6
4.3 Contraindications
Hypersensitivity to cefuroxime or to any of the excipients listed in section 6.1.
Patients with known hypersensitivity to cephalosporin antibiotics.
History of severe hypersensitivity (e.g. anaphylactic reaction) to any other type of beta-lactam antibacterial agent (penicillins, monobactams and carbapenems).
4.4 Special warnings and precautions for use
Hypersensitivity reactions
As with all beta-lactam antibacterial agents, serious and occasionally fatal hypersensitivity reactions have been reported. In case of severe hypersensitivity reactions, treatment with cefuroxime must be discontinued immediately and adequate emergency measures must be initiated.
Before beginning treatment, it should be established whether the patient has a history of severe hypersensitivity reactions to cefuroxime, to other cephalosporins or to any other type of beta-lactam agent. Caution should be used if cefuroxime is given to patients with a history of non-severe hypersensitivity to other beta-lactam agents.
Cephalosporin antibiotics may, in general, be given safely to patients who are hypersensitive to penicillins, although cross-reactions have been reported. Special care is indicated in patients who have experienced an anaphylactic reaction to penicillin.
Concurrent treatment with potent diuretics or aminoglycosides
Cephalosporin antibiotics at high dosage should be given with caution to patients receiving concurrent treatment with potent diuretics such as furosemide or aminoglycosides. Renal impairment has been reported during use of these combinations. Renal function should be monitored in the elderly and those with known pre-existing renal impairment (see section 4.2).
Overgrowth of non-susceptible microorganisms
Use of cefuroxime may result in the overgrowth of Candida. Prolonged use may also result in the overgrowth of other non-susceptible microorganisms (e.g. enterococci and Clostridium difficile), which may require interruption of treatment (see section 4.8).
Antibacterial agent–associated pseudomembranous colitis has been reported with use of cefuroxime and may range in severity from mild to life threatening. This diagnosis should be considered in patients with diarrhoea during or subsequent to the administration of cefuroxime (see section 4.8). Discontinuation of therapy with cefuroxime and the administration of specific treatment for Clostridium difficile should be considered. Medicinal products that inhibit peristalsis should not be given.
Intra-abdominal infections
Due to its spectrum of activity, cefuroxime is not suitable for the treatment of infections caused by Gram-negative non-fermenting bacteria (see section 5.1).
Interference with diagnostic tests
The development of a positive Coombs Test associated with the use of cefuroxime may interfere with cross matching of blood (see section 4.8).
Slight interference with copper reduction methods (Benedict’s, Fehling’s, Clinitest) may be observed. However, this should not lead to false-positive results, as may be experienced with some other cephalosporins.
As a false negative result may occur in the ferricyanide test, it is recommended that either the glucose oxidase or hexokinase methods are used to determine blood/plasma glucose levels in patients receiving cefuroxime sodium.
Intracameral use and eye disorders
Cefuroxime is not formulated for intracameral use. Individual cases and clusters of serious ocular adverse reactions have been reported following unapproved intracameral use of cefuroxime sodium compounded from vials approved for intravenous/intramuscular administration. These reactions included macular oedema, retinal oedema, retinal detachment, retinal toxicity, visual impairment, visual acuity reduced, vision blurred, corneal opacity and corneal oedema.
Important information about excipients
Cefuroxime powder for solution for injection and infusion contains 40.6 mg sodium per 750mg vial, equivalent to 2% of the WHO recommended maximum daily intake of 2 g sodium for an adult. This should be considered for patients who are on a controlled sodium diet.
4.5 Interaction with other medicinal products and other forms of interaction
Cefuroxime may affect the gut flora, leading to lower oestrogen reabsorption and reduced efficacy of combined oral contraceptives.
Cefuroxime is excreted by glomerular filtration and tubular secretion. Concomitant use of probenicid is not recommended. Concurrent administration of probenecid prolongs the excretion of cefuroxime and produces an elevated peak serum level.
Potential nephrotoxic drugs and loop diuretics
High-dosage treatments with cephalosporins should be carried out with caution on patients who are taking strong-acting diuretics (such as furosemide) or potential nephrotoxic preparations (such as aminoglycoside antibiotics), since impairment of renal function through such combinations cannot be ruled out.
Other Interactions
Determination of blood/plasma glucose levels: Please refer to section 4.4.
Concomitant use with oral anticoagulants may give rise to increased international normalised ratio (INR).
4.6 Fertility, pregnancy and lactation
Pregnancy
There are limited amounts of data from the use of cefuroxime in pregnant women. Studies in animals have shown no reproductive toxicity (see section 5.3). Cefuroxime should be prescribed to pregnant women only if the benefit outweighs the risk.
Cefuroxime has been shown to cross the placenta and attain therapeutic levels in amniotic fluid and cord blood after intramuscular or intravenous dose to the mother.
Breastfeeding
Cefuroxime is excreted in human milk in small quantities. Adverse reactions at therapeutic doses are not expected, although a risk of diarrhoea and fungus infection of the mucous membranes cannot be excluded. A decision must be made whether to discontinue breast-feeding or to discontinue/abstain from cefuroxime therapy taking into account the benefit of breast feeding for the child and the benefit of therapy for the woman.
Fertility
There are no data on the effects of cefuroxime sodium on fertility in humans. Reproductive studies in animals have shown no effects on fertility.
4.7 Effects on ability to drive and use machines
No studies on the effects of cefuroxime on the ability to drive and use machines have been performed. However, based on known adverse reactions, cefuroxime is unlikely to have an effect on the ability to drive and use machines.
4.8 Undesirable effects
The most common adverse reactions are neutropenia, eosinophilia, transient rise in liver enzymes or bilirubin, particularly in patients with pre-existing liver disease, but there is no evidence of harm to the liver and injection site reactions.
The frequency categories assigned to the adverse reactions below are estimates, as for most reactions suitable data for calculating incidence are not available. In addition the incidence of adverse reactions associated with cefuroxime sodium may vary according to the indication.
Data from clinical trials were used to determine the frequency of very common to rare adverse reactions. The frequencies assigned to all other adverse reactions (i.e. those occurring at <1/10,000) were mainly determined using post-marketing data, and refer to a reporting rate rather than a true frequency.
Treatment related adverse reactions, all grades, are listed below by MedDRA body system organ class, frequency and grade of severity. The following convention has been utilised for the classification of frequency: very common ≥ 1/10; common ≥ 1/100 to < 1/10, uncommon ≥ 1/1,000 to < 1/100; rare ≥ 1/10,000 to < 1/1,000; very rare < 1/10,000 and not known (cannot be estimated from the available data).

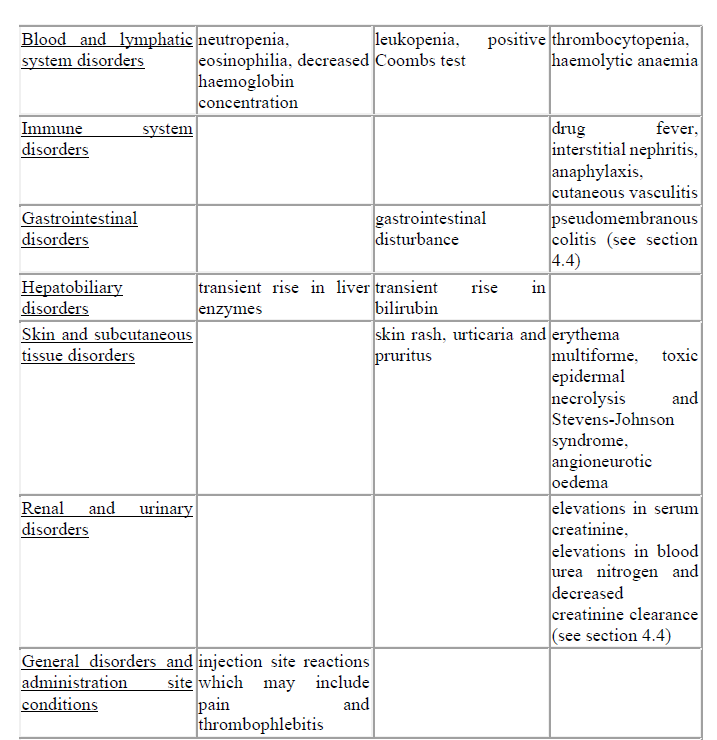
Description of selected adverse reactions
Cephalosporins as a class tend to be absorbed onto the surface of red cell membranes and react with antibodies directed against the drug to produce a positive Coombs test (which can interfere with cross matching of blood) and very rarely haemolytic anaemia.
Transient rises in serum liver enzymes or bilirubin have been observed which are usually reversible.
Pain at the intramuscular injection site is more likely at higher doses. However it is unlikely to be a cause for discontinuation of treatment.
Paediatric population
The safety profile for cefuroxime sodium in children is consistent with the profile in adults.
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via the Yellow card Scheme – Website: www.mhra.gov.uk/yellowcard or search for MHRA Yellow Card in the Google Play or Apple App Store.
4.9 Overdose
Overdose can lead to neurological sequelae including encephalopathy, convulsions and coma. Symptoms of overdose can occur if the dose is not reduced appropriately in patients with renal impairment (see sections 4.2 and 4.4).
Serum levels of cefuroxime can be reduced by haemodialysis or peritoneal dialysis.
5.0 Pharmacological properties
5.1 Pharmacodynamic properties
Pharmacotherapeutic group: antibacterials for systemic use, Second-generation cephalosporins, ATC code: J01DC02
Mechanism of action
Cefuroxime inhibits bacterial cell wall synthesis following attachment to penicillin binding proteins (PBPs). This results in the interruption of cell wall (peptidoglycan) biosynthesis, which leads to bacterial cell lysis and death.
Mechanism of resistance
Bacterial resistance to cefuroxime may be due to one or more of the following mechanisms:
• hydrolysis by beta-lactamases including (but not limited to) extended-spectrum beta-lactamases (ESBLs), and Amp-C enzymes, that may be induced or stably derepressed in certain aerobic Gram-negative bacterial species;
• reduced affinity of penicillin-binding proteins for cefuroxime;
• outer membrane impermeability, which restricts access of cefuroxime to penicillin binding proteins in Gram-negative bacteria;
• bacterial efflux pumps.
Organisms that have acquired resistance to other injectable cephalosporins are expected to be resistant to cefuroxime. Depending on the mechanism of resistance, organisms with acquired resistance to penicillins may demonstrate reduced susceptibility or resistance to cefuroxime.
Cefuroxime sodium breakpoints
Minimum inhibitory concentration (MIC) breakpoints established by the European Committee on Antimicrobial Susceptibility Testing (EUCAST) are as follows:

Microbiological susceptibility
The prevalence of acquired resistance may vary geographically and with time for selected species and local information on resistance is desirable, particularly when treating severe infections. As necessary, expert advice should be sought when the local prevalence of resistance is known and the utility of the agent in at least some types of infections is questionable.
Cefuroxime is usually active against the following microorganisms in vitro.



$ All methicillin-resistant S. aureus are resistant to cefuroxime.
In vitro the activities of cefuroxime sodium and aminoglycoside antibiotics in combination have been shown to be at least additive with occasional evidence of synergy.
5.2 Pharmacokinetic properties
Absorption
After intramuscular (IM) injection of cefuroxime to normal volunteers, the mean peak serum concentrations ranged from 27 to 35 μg/mL for a 750 mg dose and from 33 to 40 μg/mL for a 1000 mg dose, and were achieved within 30 to 60 minutes after administration. Following intravenous (IV) doses of 750 and 1500 mg, serum concentrations were approximately 50 and 100 μg/mL, respectively, at 15 minutes.
AUC and Cmax appear to increase linearly with increase in dose over the single dose range of 250 to 1000 mg following IM and IV administration. There was no evidence of accumulation of cefuroxime in the serum from normal volunteers following repeat intravenous administration of 1500 mg doses every 8 hours.
Distribution
Protein binding has been stated as 33 to 50%, depending on the methodology used. The average volume of distribution ranges from 9.3 to 15.8 L/1.73 m2 following IM or IV administration over the dosage range of 250 to 1000 mg. Concentrations of cefuroxime in excess of the minimum inhibitory levels for common pathogens can be achieved in the tonsilla, sinus tissues, bronchial mucosa, bone, pleural fluid, joint fluid, synovial fluid, interstitial fluid, bile, sputum and aqueous humour. Cefuroxime passes the blood-brain barrier when the meninges are inflamed.
Biotransformation
Cefuroxime is not metabolised.
Elimination
Cefuroxime is excreted by glomerular filtration and tubular secretion. The serum half-life after either intramuscular or intravenous administration is approximately 70 minutes. There is an almost complete recovery (85 to 90%) of unchanged cefuroxime in urine within 24 hours of administration. The majority of the cefuroxime is excreted within the first 6 hours. The average renal clearance ranges from 114 to 170 mL/min/1.73 m2 following IM or IV administration over the dosage range of 250 to 1000 mg.
Special patient populations
Gender
No differences in the pharmacokinetics of cefuroxime were observed between males and females following a single IV bolus injection of 1000 mg of cefuroxime as the sodium salt
Elderly
Following IM or IV administration, the absorption, distribution and excretion of cefuroxime in elderly patients are similar to younger patients with equivalent renal function. Because elderly patients are more likely to have decreased renal function, care should be taken in cefuroxime dose selection, and it may be useful to monitor renal function (see section 4.2).
Paediatrics
The serum half-life of cefuroxime has been shown to be substantially prolonged in neonates according to gestational age. However, in older infants (aged >3 weeks) and in children, the serum half-life of 60 to 90 minutes is similar to that observed in adults.
Renal impairment
Cefuroxime is primarily excreted by the kidneys. As with all such antibiotics, in patients with markedly impaired renal function (i.e. C1cr <20 mL/minute) it is recommended that the dosage of cefuroxime should be reduced to compensate for its slower excretion (see section 4.2). Cefuroxime is effectively removed by haemodialysis and peritoneal dialysis.
Hepatic impairment
Since cefuroxime is primarily eliminated by the kidney, hepatic dysfunction is not expected to have an effect on the pharmacokinetics of cefuroxime.
PK/PD relationship
For cephalosporins, the most important pharmacokinetic-pharmacodynamic index correlating with in vivo efficacy has been shown to be the percentage of the dosing interval (%T) that the unbound concentration remains above the minimum inhibitory concentration (MIC) of cefuroxime for individual target species (i.e. %T>MIC).
5.3 Preclinical safety data
Non-clinical data reveal no special hazard for humans based on conventional studies of safety pharmacology, repeated dose toxicity, genotoxicity and toxicity to reproduction and development. No carcinogenicity studies have been performed; however, there is no evidence to suggest carcinogenic potential.
Gamma glutamyl transpeptidase activity in rat urine is inhibited by various cephalosporins, however the level of inhibition is less with cefuroxime. This may have significance in the interference in clinical laboratory tests in humans.
6.0 Pharmaceutical particulars
6.1 List of excipients
None.
6.2 Incompatibilities
Cefuroxime is compatible with most commonly used intravenous fluids and electrolyte solutions.
The pH of 2.74% w/v sodium bicarbonate injection BP considerably affects the colour of solutions and therefore this solution is not recommended for the dilution of Cefuroxime. However, if required, for patients receiving sodium bicarbonate injection by infusion the Cefuroxime solution may be introduced into the tube of the giving set.
Cefuroxime should not be mixed in the syringe with aminoglycoside antibiotics.
In the absence of other compatibility studies, this medicinal product must not be mixed with other medicinal products apart from those listed as compatible in section 6.6.
6.3 Shelf life
Before reconstitution: 36 months
In keeping with good pharmaceutical practice, freshly constituted suspensions or solutions should be used immediately. If this is not practicable then solution may be stored at 2°C-8°C (in a refrigerator) for up to 24 hours.
6.4 Special precautions for storage
Protect from light. Before reconstitution do not store above 25°C. After reconstitution the product may be stored at 2°C-8°C (in a refrigerator) for up to 24 hours.
6.5 Nature and contents of container
Type III flint glass vial, stoppered with halobutyl closures and sealed with aluminium seals that may be combined with a polypropylene cap. Pack sizes of 1 and 10 vials. Not all pack sizes may be marketed.
6.6 Special precautions for disposal and other handling
Instructions for constitution
Table 4. Additional volumes and solution/suspension concentrations which may be useful when fractional doses are required.

*Reconstituted solution to be added to 50 or 100 ml of compatible infusion fluid (see information on compatibility, below)
** The resulting volume of the solution/suspension of cefuroxime in reconstitution medium is increased due to the displacement factor of the drug substance resulting in the listed concentrations in mg/ml.
As for all parenteral medicinal products, inspect the reconstituted solution or suspension visually for particulate matter and discoloration prior to administration.
Intramuscular injection: After addition of the specified amount of diluent for intramuscular injection, a suspension is formed.
Intravenous bolus injection or intravenous infusion: After addition of the specified amount of diluent for intravenous bolus or infusion, a clear solution is formed. The solution should only be used if the solution is clear and practically free from particles.
Solutions and suspensions range in colour from clear to yellow coloured depending on concentration, diluent and storage conditions used. When made up for intramuscular use, it becomes off-white and opaque. When made up for intravenous administration, it may be yellowish.
Compatibility
Cefuroxime sodium (5 mg/ml) in 5% w/v or 10% w/v xylitol injection may be stored for up to 24 hours at 25 °C.
Cefuroxime sodium is compatible with aqueous solutions containing up to 1% lidocaine hydrochloride.
Qvar 100 Easi-Breathe
Summary of Product Characteristics Updated 19-Oct-2017 | Teva UK Limited
Name of the medicinal product
Qvar Easi-Breathe 100 micrograms per actuation pressurised inhalation solution
Qualitative and quantitative composition
Beclometasone Dipropionate 100 micrograms per metered (ex-valve) dose.
For the full list of excipients, see section 6.1.
Pharmaceutical form
Pressurised inhalation, solution.
A colourless solution in a pressurised aluminium canister fitted with a metering valve and an actuator.
Qvar Easi-Breathe contains a propellant, which does not contain any chlorofluorocarbons (CFCs).
Clinical particulars
4.1 Therapeutic indications
Prophylactic management of mild, moderate or severe asthma.
4.2 Posology and method of administration
Posology
Qvar Easi-Breathe is for inhalation use only.
Patients should be instructed in the proper use of their inhaler, including rinsing out their mouth with water after use..
NOTE: The recommended total daily dose of Qvar Easi-Breathe is lower than that for current beclometasone dipropionate containing products and should be adjusted to the needs of the individual patient.
ADULT STARTING AND MAINTENANCE DOSE:
It is important to gain control of asthma symptoms and optimise pulmonary function as soon as possible. When patients’ symptoms remain under satisfactory control, the dose should be titrated to the lowest dose at which effective control of asthma is maintained.
To be effective inhaled Qvar Easi-Breathe must be used on a regular basis even when patients are asymptomatic.
THERAPY IN NEW PATIENTS SHOULD BE INITIATED AT THE FOLLOWING
Mild asthma: 100 to 200 micrograms per day in two divided doses.
Moderate asthma: 200 to 400 micrograms per day in two divided doses.
Severe asthma: 400 to 800 micrograms per day in two divided doses.
Patients on budesonide inhalers may be transferred to Qvar as described below.
The general approach to switching patients to Qvar involves two steps as detailed below. Specific guidance on switching well-controlled and poorly-controlled (symptomatic) patients is given below the table.
Step 1: Consider the dose of budesonide-containing inhalers appropriate to the patient’s current condition.
Step 2: Convert the budesonide inhaler dose to the Qvar dose according to the table below.
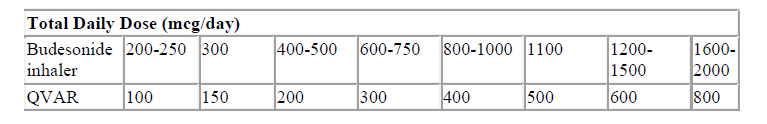
Patients with well-controlled asthma using budesonide inhaler products should be switched to Qvar at a dose in accordance with the table above.
For example:
Patients on 2 puffs twice daily of budesonide 100 micrograms would change to 2 puffs twice daily of Qvar 50 micrograms.
Patients with poorly-controlled asthma may be switched from budesonide inhaler products to Qvar at the same microgram for microgram dose up to 800 micrograms daily.
Alternatively the patient’s current budesonide inhaler dose can be doubled and this dose can be converted to the Qvar dose according to the table above.
Patients on fluticasone inhalers may be transferred to the same total daily dose of Qvar up to 800 micrograms daily.
Once transferred to Qvar Easi-Breathe the dose should be adjusted to meet the needs of the individual patient.
The maximum recommended dose is 800 micrograms per day in divided doses.
The same total daily dose in micrograms from either Qvar Easi-Breathe 50 (a lower strength) or Qvar Easi-Breathe 100 Inhaler provides the same clinical effect.
Patients should be instructed in the proper use of their inhaler, including rinsing out their mouth with water after use. Patients should be advised that Qvar Easi-Breathe may have a different taste and feel than a CFC inhaler.
Paediatric population
There are no data to date on Qvar Easi-Breathe in children under 12 years of age, hence no definitive dosage recommendation can be made.
Special patient groups
No special dosage recommendations are made for elderly or patients with hepatic or renal impairment.
Method of administration
The aerosol spray is inhaled through the mouth into the lungs. The inhaler should be primed by firing two shots into the air before first use or if the inhaler has not been used for a period of two weeks or longer.
After removal of the cap the inhaler mouthpiece should be placed in the mouth with the lips closed around it. The patient should breathe in slowly and deeply through the mouthpiece. They should be advised not to stop breathing when the inhaler delivers the dose into their mouth but carry on until they have taken a deep breath to ensure optimal delivery of the product.
For normal hygiene, the mouthpiece of the inhaler should be cleaned weekly with a clean dry tissue or cloth. The inhaler should not be washed or immersed in water at any time.
Full instructions for use are given in the Patient Information Leaflet, which should be read carefully by the patient before use.
Qvar Easi-Breathe delivers a consistent dose
- whether or not the canister is shaken by the patient
- without the need for the patient to wait between individual actuations
- regardless of storage orientation or periods without use of up to 14 days
- at temperatures as low as -10°C.
4.3 Contraindications
Hypersensitivity to the active substance or to any of the excipients listed in section 6.1.
4.4 Special warnings and precautions for use
Patients should be properly instructed on the use of the inhaler to ensure that the drug reaches the target areas within the lungs. To be effective, Qvar Easi-Breathe must be used by patients on a regular basis, even when patients do not have asthma symptoms. When symptoms are controlled, maintenance Qvar Easi-Breathe therapy should be reduced in a stepwise manner to the minimum effective dose. Inhaled steroid treatment should not be stopped abruptly.
Patients with asthma are at risk of acute attacks and should have regular assessments of their asthma control including pulmonary function tests.
Qvar Easi-Breathe is not indicated for the immediate relief of asthma attacks. Patients therefore need to have relief medication (inhaled short-acting bronchodilator) available for such circumstances.
Severe asthma requires regular medical assessment, including lung-function testing, as there is a risk of severe attacks and even death. Patients should be instructed to seek medical attention if short-acting relief bronchodilator treatment becomes less effective, or more inhalations than usual are required as this may indicate deterioration of asthma control. If this occurs, patients should be assessed and the need for increased anti-inflammatory therapy considered (eg. higher doses of inhaled corticosteroid or a course of oral corticosteroid)
Severe asthma exacerbations should be managed in the usual way i.e. by increasing the dose of inhaled beclometasone dipropionate, giving a systemic steroid if necessary, and/or an appropriate antibiotic if there is an infection, together with β-agonist therapy.
Treatment with Qvar should not be stopped abruptly.
However, systemic effects of inhaled corticosteroids may occur, particularly at high doses prescribed for prolonged periods. These effects are much less likely to occur than with oral corticosteroids. Possible systemic effects include Cushing’s syndrome, Cushingoid features, adrenal suppression, growth retardation in children and adolescents, decrease in bone mineral density, cataract, glaucoma, blurred vision, and more rarely, a range of psychological or behavioural effects including psychomotor hyperactivity, sleep disorders, anxiety, depression or aggression (particularly in children). It is important, therefore, that the dose of inhaled corticosteroid is titrated to the lowest dose at which effective control of asthma is maintained.
It is recommended that the height of children receiving prolonged treatment with inhaled corticosteroids is regularly monitored. If growth is slowed, therapy should be reviewed with the aim of reducing the dose of inhaled corticosteroid, if possible, to the lowest dose at which effective control of asthma is maintained. In addition, consideration should be given to referring the patient to a paediatric respiratory specialist.
Prolonged treatment with high doses of inhaled corticosteroids, particularly higher than the recommended doses, may result in clinically significant adrenal suppression.
Additional systemic corticosteroid cover should be considered during periods of stress or elective surgery.
Patients who have received systemic steroids for long periods of time or at high doses, or both, need special care and subsequent management when being transferred to inhaled steroid therapy. Patients should have stable asthma before being given inhaled steroids in addition to the usual maintenance dose of systemic steroid. Withdrawal of systemic steroids should be gradual, starting about seven days after the introduction of Qvar Easi-Breathe therapy. For daily oral doses of prednisolone of 10mg or less, dose reduction in 1mg steps, at intervals of not less than one week is recommended. For patients on daily maintenance doses of oral prednisolone greater than 10mg, larger weekly reductions in the dose might be acceptable. The dose reduction scheme should be chosen to correlate with the magnitude of the maintenance systemic steroid dose.
As recovery from impaired adrenocortical function, caused by prolonged systemic steroid therapy is slow, adrenocortical function should be monitored regularly.
Patients should be advised that they may feel unwell in a non-specific way during systemic steroid withdrawal despite maintenance of, or even improved respiratory function. Patients should be advised to persevere with their inhaled product and to continue withdrawal of systemic steroids, even if feeling unwell, unless there is evidence of HPA axis suppression.
Patients weaned off oral steroids whose adrenocortical function is impaired should carry a steroid warning card indicating that they may need supplementary systemic steroids during periods of stress, eg. worsening asthma attacks, chest infections, major intercurrent illness, surgery, trauma, etc.
Discontinuation of systemic steroids may also cause exacerbation of allergic diseases such as atopic eczema and rhinitis. These should be treated as required with topical therapy, including corticosteroids and/or antihistamines.
Like other corticosteroids, caution is necessary in patients with active or latent pulmonary tuberculosis.
Patients should be advised to seek medical attention for review of maintenance Qvar Easi-Breathe therapy if peak flow falls, symptoms worsen or if the short-acting bronchodilator becomes less effective and increased inhalations are required. This may indicate worsening asthma.
Most patients can be successfully transferred to inhaled steroids with maintenance of good respiratory function, but special care is necessary for the first few months after the transfer, until the hypothalamic-pituitary-adrenal (HPA) system has sufficiently recovered to enable the patient to cope with stressful emergencies such as trauma, surgery or serious infections. Patients should, therefore, carry a steroid warning card to indicate the possible need to re-instate systemic steroid therapy rapidly during periods of stress or where airways obstruction or mucus significantly compromises the inhaled route of administration. In addition, it may be advisable to provide such patients with a supply of corticosteroid tablets to use in these circumstances. The dose of inhaled steroids should be increased at this time and then gradually reduced to the maintenance level after the systemic steroid has been discontinued.
Beclometasone dipropionate, like other inhaled steroids, is absorbed into the systemic circulation from the lungs. Beclometasone dipropionate and its metabolites may exert detectable suppression of adrenal function. Within the dose range 100-800 micrograms daily, clinical studies with Qvar Easi-Breathe have demonstrated mean values for adrenal function and responsiveness within the normal range.
Visual disturbance may be reported with systemic and topical corticosteroid use. If a patient presents with symptoms such as blurred vision or other visual disturbances, the patient should be considered for referral to an ophthalmologist for evaluation of possible causes which may include cataract, glaucoma or rare diseases such as central serous chorioretinopathy (CSCR) which have been reported after use of systemic and topical corticosteroids.
Patients should be advised that this product contains small amounts of ethanol. At the normal doses, the amounts of ethanol are negligible and do not pose a risk to patients (see section 4.5).
4.5 Interaction with other medicinal products and other forms of interaction
Qvar Easi-Breathe contains a small amount of ethanol. There is a theoretical potential for interaction in particularly sensitive patients taking disulfiram or metronidazole.
Beclometasone is less dependent on CYP3A metabolism than some other corticosteroids, and in general interactions are unlikely; however the possibility of systemic effects with concomitant use of strong CYP3A inhibitors (e.g. ritonavir, cobicistat) cannot be excluded, and therefore caution and appropriate monitoring is advised with the use of such agents.
4.6 Fertility, pregnancy and lactation
The potential risk of this product for humans is unknown.
Qvar Easi-Breathe
There is no experience of this product in pregnancy and lactation in humans, therefore the product should only be used if the expected benefits to the mother are thought to outweigh any potential risk to the foetus or neonate
Beclometasone dipropionate
Pregnancy
There is inadequate evidence of safety in human pregnancy. Administration of corticosteroids to pregnant animals can cause abnormalities of foetal development including cleft palate and intra-uterine growth retardation. There may therefore, be a risk of such effects in the human foetus. It should be noted, however, that the foetal changes in animals occur after relatively high systemic exposure. Beclometasone dipropionate is delivered directly to the lungs by the inhaled route and so avoids the high level of exposure that occurs when corticosteroids are given by systemic routes.
The use of beclometasone dipropionate in pregnancy requires that the possible benefits of the drug be weighed against the possible hazards. The drug has been in widespread use for many years without apparent ill consequence.
Breast-feeding
No specific studies examining the transfer of beclometasone dipropionate into the milk of lactating animals have been performed. It is probable that beclometasone dipropionate is excreted in milk. However, given the relatively low doses used by the inhalation route, the levels are likely to be low. In mothers breast feeding their baby the therapeutic benefits of the drug should be weighed against the potential hazards to mother and baby.
There is no experience with or evidence of safety of propellant HFA 134a in human pregnancy or lactation. However, studies on the effect of HFA 134a on reproductive function and embryofoetal development in animals have revealed no clinically relevant adverse effects.
4.7 Effects on ability to drive and use machines
Not relevant.
4.8 Undesirable effects
A serious hypersensitivity reaction including oedema of the eye, face, lips and throat (angioedema) has been reported rarely.
As with other inhaled therapy, paradoxical bronchospasm may occur after dosing. Immediate treatment with a short-acting bronchodilator should be initiated, Qvar should be discontinued immediately and an alternate prophylactic treatment introduced.
Systemic effects of inhaled corticosteroids may occur, particularly with high doses prescribed for prolonged periods. These include adrenal suppression, growth retardation in children, decrease in bone mineral density and the occurrence of cataract and glaucoma.
Commonly, when taking Qvar, hoarseness and candidiasis of the throat and mouth may occur. To reduce the risk of hoarseness and candida infection, patients are advised to rinse their mouth after using their inhaler.
Based on the MedDra system organ class and frequencies, adverse events are listed in the table below according to the following frequency estimate: very common (≥ 1/10); common (≥1/100 to <1/10); Uncommon (≥1/1,000 to <1/100); rare (≥1/10,000 to <1/1,000); very rare (<1/10,000), not known (cannot be estimated from the available data).

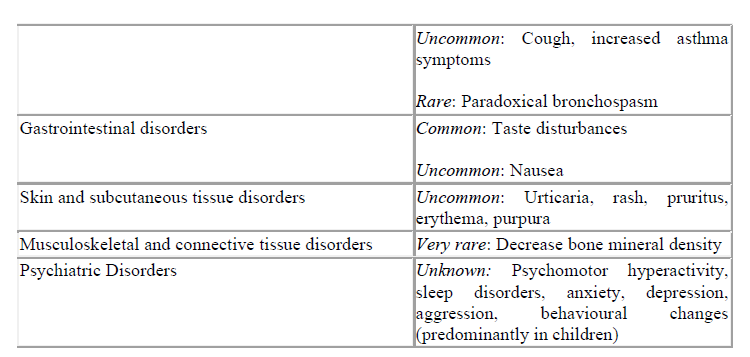
*Systemic reactions are a possible response to inhaled corticosteroids, especially when a high dose is prescribed for a prolonged time (see section 4.4 Special warnings and precautions for use).
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via the Yellow Card Scheme at: www.mhra.gov.uk/yellowcard
4.9 Overdose
Acute overdosage is unlikely to cause problems. The only harmful effect that follows inhalation of large amounts of the drug over a short time period is suppression of HPA function. Specific emergency action need not be taken. Treatment with Qvar Easi-Breathe should be continued at the recommended dose to control the asthma; HPA function recovers in a day or two.
If excessive doses of beclometasone dipropionate were taken over a prolonged period a degree of atrophy of the adrenal cortex could occur in addition to HPA suppression. In this event the patient should be treated as steroid dependent and transferred to a suitable maintenance dose of a systemic steroid such as prednisolone. Once the condition is stabilised, the patient should be returned to Qvar Easi-Breathe by the method described above in Section 4.4.
5.0 Pharmacological properties
5.1 Pharmacodynamic properties
Pharmacotherapeutic group: Glucocorticoids, ATC code: R03BA01
Qvar Easi-Breathe contains beclometasone dipropionate in solution in propellant HFA-134a resulting in an extra fine aerosol. The aerosol droplets are on average much smaller than the beclometasone dipropionate particles delivered by CFC-suspension formulations or dry powder formulations of beclometasone dipropionate. The extra fine particle fraction will be 60% ± 20% of the drug particles ≤ 3.3 microns per shot, ex-actuator.
Radiolabelled deposition studies in adults with mild asthma have demonstrated that the majority of drug (> 55% ex-actuator) is deposited in the lung and a small amount (< 35% ex-actuator) is deposited in the oropharynx. These studies were performed with Qvar Aerosol. Qvar Aerosol is a ‘press and breathe’ inhaler, whereas Qvar Easi-Breathe is a breath-activated inhaler.
Inhaled beclometasone dipropionate is now well established in the management of asthma. It is a synthetic glucocorticoid and exerts a topical, anti-inflammatory effect on the lungs, with fewer systemic effects than oral corticosteroids.
Comparative clinical studies of Qvar aerosol have demonstrated that asthma patients achieve equivalent pulmonary function and control of symptoms with Qvar aerosol at lower total daily doses than CFC containing beclometasone dipropionate aerosol inhalers.
Pharmacodynamic studies in patients with mild asthma given Qvar aerosol for 14 days, have shown that there is a linear correlation among urinary free cortisol suppression, dose administered, and serum total-beclometasone levels obtained. At a daily dose of 800 micrograms Qvar aerosol, suppression of urinary free cortisol was comparable with that observed with the same daily dose of CFC containing beclometasone dipropionate, indicating a wider safety margin, as Qvar Easi-Breathe is administered at lower doses than the CFC product.
5.2 Pharmacokinetic properties
The pharmacokinetic profile of Qvar aerosol (an equivalent inhaler) shows that the peak serum concentration for total- beclometasone (BOH) (total of any beclometasone OH and beclometasone dipropionate or monopropionate hydrolysed to beclometasone OH) or after single and multiple doses is achieved after 30 minutes.
The value at the peak is approximately 2 nanograms/ml after a total daily dose of 800 micrograms and the serum levels after 100, 200 and 400 micrograms are proportional. The principal route of elimination of beclometasone dipropionate and its several metabolites is in the faeces. Between 10% and 15% of an orally administered dose is excreted in the urine, as both conjugated and free metabolites of the drug.
In both single dose and multiple dose pharmacokinetic studies of Qvar aerosol, a dose of 200 micrograms of Qvar aerosol achieved comparable total-BOH levels, as a dose of 400 micrograms of CFC containing beclometasone dipropionate Aerosol. This provided the scientific rationale for investigating lower total daily doses of Qvar aerosol to achieve the same clinical effect.
Pharmacokinetic studies with Qvar Easi-Breathe have not been carried out in any special populations.
Section Content
About Instructor

Section Includes
- 1 Mock Exam